![]()
![]()
![]()
Using ESMFold2#
ESMFold2 is the latest generation of the ESM structure-prediction family (Biohub/esm). Unlike first-generation ESMFold, it predicts full biomolecular complexes (multiple protein chains, nucleic acids, and small-molecule ligands) with a diffusion-based decoder, and can optionally condition on a multiple sequence alignment (MSA) for improved accuracy.
Two variants are available:
ESMFold2Model(session.fold.esmfold2): the full model; accepts an optional MSA per protein chain.ESMFold2FastModel(session.fold.esmfold2_fast): a single-sequence variant for fast predictions without an MSA. It uses half the folding layers of the full model.
Throughout this guide we use the mature 99-residue HIV-1 protease, a classic obligate homodimer: two identical chains pair up to form the functional enzyme, with a single active site at their interface. It is a long-standing target of antiretroviral drugs such as ritonavir, which we fold alongside it later.
What you need before getting started#
Make sure you have an active OpenProtein session, then import the classes used to assemble a complex and define the protease sequence:
[1]:
import openprotein
from openprotein.molecules import Complex, Protein, Ligand
# Login to your session
session = openprotein.connect()
# Mature HIV-1 protease (99 residues), a classic obligate homodimer
sequence = (
"PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI"
"GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF"
)
Getting the model#
Create the model object and inspect its fold signature:
[2]:
esmfold2 = session.fold.esmfold2
help(esmfold2.fold)
Help on method fold in module openprotein.fold.esmfold2:
fold(
sequences: Sequence[Complex | Protein | str | bytes] | MSAFuture,
diffusion_samples: int = 1,
num_recycles: int = 3,
num_steps: int = 100,
seed: int | None = None,
**_
) -> FoldResultFuture method of openprotein.fold.esmfold2.ESMFold2Model instance
Request structure prediction with ESMFold2.
Parameters
----------
sequences : Sequence[Complex | Protein | str | bytes] | MSAFuture
List of complexes to fold. `Protein` objects must be tagged with
an `msa`, which can be `Protein.single_sequence_mode` for single
sequence mode. Alternatively, supply an `MSAFuture` to use all
query sequences as a multimer.
diffusion_samples : int
Number of diffusion samples to use.
num_recycles : int
Number of recycling steps to use.
num_steps : int
Number of sampling steps to use.
seed : int | None
Seed for the diffusion sampler.
Returns
-------
FoldResultFuture
Future for the folding result.
Folding a complex#
The functional protease is a homodimer: two identical chains whose catalytic site forms at their interface, so we fold both chains together as a Complex. Because the two chains are identical, we build a single Protein, tag it with Protein.single_sequence_mode to fold without an MSA, and place it in both chain slots.
The runtime hyperparameters trade speed for accuracy:
num_recycles: how many times the trunk refines its representation.num_steps: diffusion sampling steps. The default is 100, but fewer often suffice with little loss in quality; we use 50.diffusion_samples: number of independent structures drawn per input.seed: makes a run reproducible.
Submitting a fold returns a FoldResultFuture; wait for it with wait_until_done():
[3]:
protease = Protein(sequence)
protease.msa = Protein.single_sequence_mode
dimer = Complex(chains={"A": protease, "B": protease})
dimer_future = esmfold2.fold(
[dimer],
num_recycles=3, # trunk recycling iterations
num_steps=50, # diffusion sampling steps (default 100)
diffusion_samples=1, # structures drawn per input
seed=0,
)
dimer_future.wait_until_done(verbose=True, timeout=900)
Waiting: 100%|██████████| 100/100 [00:00<00:00, 1559.45it/s, status=SUCCESS]
[3]:
True
Retrieving and visualizing the structure#
Getting the structure#
Fetch results with get(). It returns one Structure per input, and each Structure holds one Complex per diffusion sample:
[4]:
structure = dimer_future.get()[0]
predicted = structure[0] # first diffusion sample
print("Predicted structure:", structure)
print("Chain A:", predicted.get_protein("A").sequence.decode())
print("Chain B:", predicted.get_protein("B").sequence.decode())
Predicted structure: <openprotein.molecules.structure.Structure object at 0x17703b230>
Chain A: PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
Chain B: PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
Visualizing the structure#
Render the prediction with molviewspec, coloring each chain separately so the two halves of the dimer are easy to tell apart:
[5]:
%pip install molviewspec
from molviewspec import create_builder
def display_structure(structure_string):
builder = create_builder()
(
builder.download(url="mystructure.cif")
.parse(format="mmcif")
.model_structure()
.component()
.representation()
.color_from_source(
schema="atom",
category_name="atom_site",
field_name="auth_asym_id",
palette={
"kind": "categorical", # color by chain
"colors": ["blue", "red", "green", "orange"],
"mode": "ordinal",
},
)
)
return builder.molstar_notebook(
data={"mystructure.cif": structure_string}, width=500, height=400
)
display_structure(structure.to_string(format="cif"))
Requirement already satisfied: molviewspec in /Users/ttruong/code/openprotein-docs/.pixi/envs/dev-nb/lib/python3.14/site-packages (1.8.1)
Requirement already satisfied: pydantic<3,>=1 in /Users/ttruong/code/openprotein-docs/.pixi/envs/dev-nb/lib/python3.14/site-packages (from molviewspec) (2.12.5)
Requirement already satisfied: annotated-types>=0.6.0 in /Users/ttruong/code/openprotein-docs/.pixi/envs/dev-nb/lib/python3.14/site-packages (from pydantic<3,>=1->molviewspec) (0.7.0)
Requirement already satisfied: pydantic-core==2.41.5 in /Users/ttruong/code/openprotein-docs/.pixi/envs/dev-nb/lib/python3.14/site-packages (from pydantic<3,>=1->molviewspec) (2.41.5)
Requirement already satisfied: typing-extensions>=4.14.1 in /Users/ttruong/code/openprotein-docs/.pixi/envs/dev-nb/lib/python3.14/site-packages (from pydantic<3,>=1->molviewspec) (4.15.0)
Requirement already satisfied: typing-inspection>=0.4.2 in /Users/ttruong/code/openprotein-docs/.pixi/envs/dev-nb/lib/python3.14/site-packages (from pydantic<3,>=1->molviewspec) (0.4.2)
Note: you may need to restart the kernel to use updated packages.
Assessing prediction confidence#
ESMFold2 reports per-sample confidence via get_confidence(), returning one ESMFold2Confidence per diffusion sample. For a multi-chain complex the scores describe both the whole assembly and the relationships between chains:
pTM (predicted TM-score): global accuracy of the overall fold (0–1, higher is better).
ipTM (interface pTM): accuracy of the relative placement of the chains; the key score for whether the interface is right.
complex pLDDT: mean per-residue confidence across the complex.
per-chain pTM (
chains_ptm): pTM computed for each chain alone.pairwise chain ipTM (
pair_chains_iptm): ipTM for each ordered pair of chains, i.e. how confidently chain i is placed relative to chain j.
A single chain has no interface, so ipTM and the pairwise table only become meaningful once you fold two or more chains, as we do here:
[6]:
confidence = dimer_future.get_confidence()[0][0] # [input][diffusion sample]
print(f"pTM: {confidence.ptm:.3f}")
print(f"ipTM: {confidence.iptm:.3f}")
print(f"complex pLDDT: {confidence.complex_plddt:.3f}")
print("per-chain pTM: " + ", ".join(
f"{cid}={val:.3f}" for cid, val in confidence.chains_ptm.items()
))
# Pairwise chain ipTM, as a labeled grid (row placed relative to column)
pair = confidence.pair_chains_iptm
chain_ids = list(pair.keys())
print("\npairwise chain ipTM:")
print(" " + "".join(f"{c:>8}" for c in chain_ids))
for i in chain_ids:
row = "".join(f"{pair[i][j]:8.3f}" for j in chain_ids)
print(f"{i:>5} {row}")
pTM: 0.957
ipTM: 0.950
complex pLDDT: 0.940
per-chain pTM: 0=0.886, 1=0.886
pairwise chain ipTM:
0 1
0 0.886 0.797
1 0.797 0.886
Visualizing PAE and pLDDT#
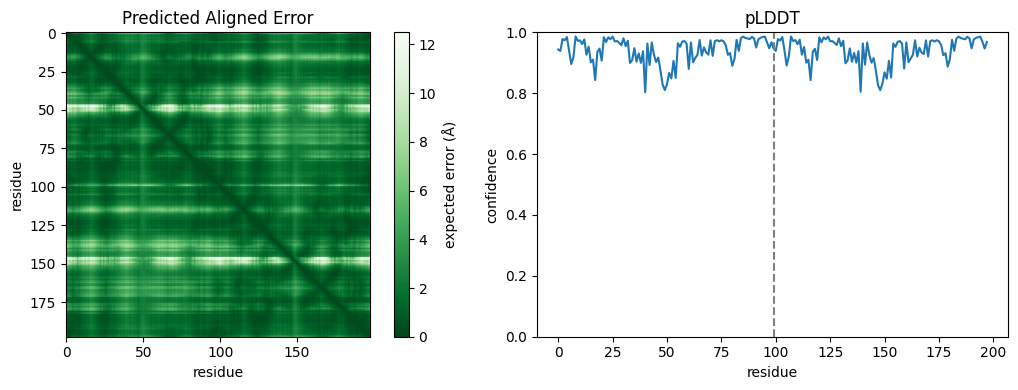
get_pae() returns the Predicted Aligned Error: an N×N matrix (in ångströms) whose entry (i, j) is the expected error in residue j’s position when the structure is aligned on residue i. Confident off-diagonal blocks between the two chains signal a well-predicted interface. get_plddt() returns the per-residue pLDDT. Both arrive with a leading diffusion-sample axis, so we take the first sample:
[7]:
import matplotlib.pyplot as plt
pae = dimer_future.get_pae()[0][0] # (samples, N, N) -> first sample
plddt = dimer_future.get_plddt()[0][0] # (samples, N) -> first sample
fig, (ax_pae, ax_plddt) = plt.subplots(1, 2, figsize=(11, 4))
im = ax_pae.imshow(pae, cmap="Greens_r", vmin=0)
ax_pae.set(title="Predicted Aligned Error", xlabel="residue", ylabel="residue")
fig.colorbar(im, ax=ax_pae, label="expected error (Å)")
ax_plddt.plot(plddt)
ax_plddt.set(title="pLDDT", xlabel="residue", ylabel="confidence", ylim=(0, 1))
ax_plddt.axvline(len(plddt) // 2, ls="--", color="grey") # chain A / B boundary
plt.tight_layout()
plt.show()
Conditioning on an MSA#
The folds so far used single-sequence mode, which already gives ESMFold2 strong accuracy. Conditioning each protein chain on a multiple sequence alignment can improve it further; the full esmfold2 model supports this as an option. Build an MSA from the query sequence with session.align.create_msa and assign it to the chain’s msa attribute. Both chains are identical, so we attach the MSA to a single Protein and place it in both chain
slots:
[8]:
msa = session.align.create_msa(sequence.encode())
msa_protease = Protein(sequence)
msa_protease.msa = msa
msa_dimer = Complex(chains={"A": msa_protease, "B": msa_protease})
msa_future = esmfold2.fold([msa_dimer], num_steps=50, seed=0)
msa_future.wait_until_done(verbose=True, timeout=900)
Waiting: 100%|██████████| 100/100 [04:00<00:00, 2.40s/it, status=SUCCESS]
[8]:
True
Folding with ligands, DNA, and RNA#
Because ESMFold2 predicts whole complexes, a Complex can mix protein chains with small-molecule ligands, DNA, and RNA. Here we co-fold the protease dimer with ritonavir, an antiretroviral drug designed to inhibit it, supplied as a Ligand by its Chemical Component Dictionary (CCD) code:
[9]:
ligand_dimer = Complex(chains={
"A": protease,
"B": protease,
"L": Ligand(ccd="RIT"), # ritonavir, by CCD code
# ...a ligand can also be given a SMILES string: Ligand(smiles=...)
})
ligand_future = esmfold2.fold([ligand_dimer], num_steps=50, seed=0)
ligand_future.wait_until_done(verbose=True, timeout=900)
Waiting: 100%|██████████| 100/100 [00:00<00:00, 1674.06it/s, status=SUCCESS]
[9]:
True
DNA and RNA chains follow the same pattern: build them from a nucleotide sequence and add them under their own chain id. The protease does not bind nucleic acids, but the API looks like this:
from openprotein.molecules import DNA, RNA
nucleic_complex = Complex(chains={
"A": protease,
"D": DNA("GGAATTCC"), # a DNA chain
"R": RNA("GGGAGG"), # an RNA chain
})
Using ESMFold2-Fast#
esmfold2_fast is a lighter-weight, faster variant that uses half the folding layers, making it well-suited to high-throughput screens where you need to fold many sequences quickly. It takes the same inputs and hyperparameters, but every protein chain must use Protein.single_sequence_mode; it rejects chains that carry an MSA:
[10]:
esmfold2_fast = session.fold.esmfold2_fast
fast_chain = Protein(sequence)
fast_chain.msa = Protein.single_sequence_mode
fast_future = esmfold2_fast.fold([fast_chain], num_steps=50, seed=0)
fast_future.wait_until_done(verbose=True, timeout=900)
Waiting: 100%|██████████| 100/100 [00:00<00:00, 1453.59it/s, status=SUCCESS]
[10]:
True
A note on ESMFold (first generation)#
First-generation ESMFold remains available via session.fold.esmfold for quick single-sequence protein predictions. It does not support ligands, nucleic acids, or MSA conditioning, but it is a fast option when you only need to fold protein chains:
[11]:
esmfold = session.fold.esmfold
esm_structure = esmfold.fold([sequence.encode()]).get()[0]
print("ESMFold (v1) prediction:", esm_structure)
ESMFold (v1) prediction: <openprotein.molecules.structure.Structure object at 0x30a0de710>
Next steps#
Save a prediction for later, or compare ESMFold2 against another predictor such as AlphaFold2, Boltz, or Protenix-v2:
[12]:
with open("esmfold2_prediction.cif", "w") as f:
f.write(structure.to_string(format="cif"))